
The VA PGCoE applies expertise in interconnected areas including biomedical, basic, and clinical research; development of genomic technologies including metagenomics; and application of clinical and Public Health (PH) sequencing quality standards as it relates to infectious diseases with the goal of improving the translation of pathogen genomic technologies. The VA PGCoE acts as an incubator to develop, optimize, and standardize methods to expedite PH adoption in partnership with others to address limitations preventing the broader application of genomics for PH action. The projects establish and expand genomics to inform epidemiology and PH within Virginia and the United States.
Landscape Analysis
The VA PGCoE is engaged in several landscape analysis efforts to document and characterize Genomic Epidemiology data, tools, processes, and systems currently in use within the center and across the PGCoE network. This information is used to identify opportunities for enhancements in efficiency, analysis, and outbreak response. To learn more about the specific efforts of the VA PGCoE, click on the plus sign (+) to expand the list of items below:
NGS Wet Lab Processes in Public Health Laboratories (PHLs)
Background: To identify the capabilities and gaps in knowledge and skills of Public Health Laboratories (PHLs), a landscape analysis will be undertaken of wet-laboratory methods and technologies. This will include the execution of surveys, focus group interviews, and assessments of needs for prioritization within the U.S. PH system. The data will be used to refine and direct grant activities in later years.
Purpose: Perform a landscape analysis of wet lab method gaps, needs, and opportunities for the development of innovative genomic technologies. This assessment will identify limitations and enable a broader assessment of current wet-lab methodologies in PHLs.
Outcomes:
- Reduce “blind-spots” in PH by understanding gaps and variability in wet lab approaches.
- Improve the translation of research-based approaches to PHLs, addressing projects specifically proposed by the VA PGCoE and extended to other priorities of the CDC.
- Address limitations for the adoption of additional sequencing applications by capitalizing on academic expertise for method consultation, development, and refinement.
- Contribute information gained through these activities to the CDC/PGCoE Network Roadmap for pathogen genomics and genomic epidemiology in public health.
Bioinformatics Capabilities across the Mid-Atlantic Region
Background: The VA PGCoE will assist in understanding the regional strengths and approaches to bioinformatics. Despite the widespread use of bioinformatics among PHL, there has not been a comprehensive assessment of bioinformatic methodologies and approaches available. Illuminating unique computing approaches and how bioinformatics tasks are performed will identify gaps, needs, and opportunities for bioinformatics development and standardization.
Purpose: Landscape analysis of bioinformatics in PHLs is crucial to identify gaps and future opportunities and to effectively and efficiently address needs that become apparent across multiple labs to employ standard approaches so data and analysis can more easily be compared. In addition, a bioinformatic landscape analysis may highlight the disparities in education and training among the public health workforce and can be used to more equitably educate scientists so that there is a baseline of knowledge and capability that is standard across all PHLs. It is important to note that the results of the landscape analysis may significantly alter the subsequent detailed plans in the project in order to address the identified needs. This type of flexibility is crucial in order for the PGCoE to be responsive to PHL and other stakeholder needs.
Outcomes:
- Using surveys, questionnaires, individual interviews, calls, and other data-gathering methods as deemed necessary, gain a baseline-level knowledge of the bioinformatic capacity across the regional PHLs.
- Produce a report that identifies common areas of weakness, or lack of bioinformatic education in the public health workforce.
- Highlight variability across algorithms and code to develop standardized approaches.
Genomic Surveillance
Background: Pathogen whole genome sequencing (WGS) surveillance data can inform public health by establishing a baseline of the molecular epidemiology and over time can identify patterns or clusters that were previously invisible. There needs to be an accounting for ongoing projects to understand the next steps in more prioritized and effective deployment of genomic surveillance for public health.
Purpose: The Virginia PGCoE will assess current genomic surveillance efforts across the Mid-Atlantic Region to assess what is occurring in our region that may be leveraged and synergistic across the states. This can also identify gaps and barriers in our region and how regional work can synergize with the wider PGCoE.
Outcomes:
- Perform a detailed survey of ongoing genomic surveillance to regional state PHL.
- Compare and contrast the pathogens, focus, methodology, reporting structure, and data transfer across the region.
Field Applications
Background: There are many successful and innovative approaches to pathogen genomics. However, genomics cannot help to improve public health if it is not connected with actionable data in the hands of epidemiologists who can drive public health interventions.
Purpose: Using SARS-CoV-2 surveillance as a use case will help to better understand gaps in field applications from sequencing networks and genomics and inform public health action.
Outcomes: Provide a landscape analysis report after nationwide interview of state public health laboratory leadership as well as state public health department leadership to understand SARS-CoV-2 genomic applications and use of this data.
Wet Laboratory Methods
Wet laboratory methods refer to analyses that are performed using physical biological samples, as opposed to analyses performed using data and computational methods. Genome sequencing of pathogens is a key component of Genomic Epidemiology. In the VA PGCoE, efforts are underway to improve efficiency, throughput, and accuracy of sequencing of pathogens isolated from people and the environment. Click on the plus sign (+) to expand the list of items below to learn more:
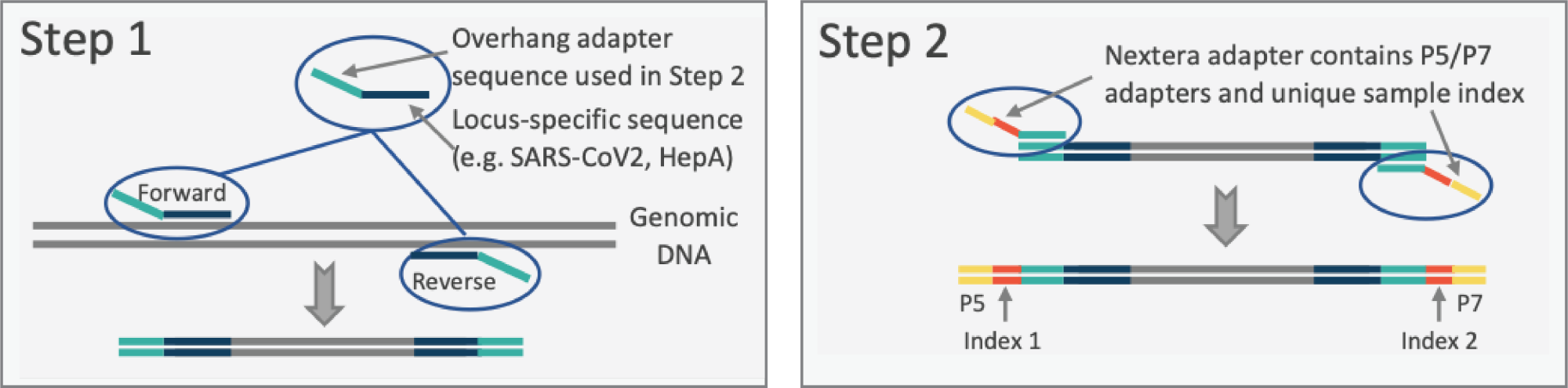
Background: Two approaches are proposed for viral genome sequencing: 1) a novel sequencing strategy — Pathogenic Virus Amplification and Sequencing Protocol (PVASP; see figures); and 2) metagenomic/metatranscriptomic sequence (MGS/MTS) profiling. The former targets one pathogen at a time for which a good reference sequence is available. The latter are ‘pan-microbial’, providing classifications of viruses, bacteria, fungi & parasites, AMR genes, and gene expression profiles that provide metabolic/pathogenic activities of these agents.
Purpose:
- Amplification-based viral pathogen genome sequencing. To optimize and apply the PVASP, originally adopted from rapid microbiome profiling and applied to sequencing of the genomes of SARS-CoV-2 (SC2) isolates, to viral disease outbreaks. Once optimized, the PVASP will be applicable to any viral pathogen, either RNA- or DNA-based. We have proposed to use Hepatitis A Virus (HAV) as a testbed for establishing and optimizing this pathogen-agnostic protocol. Hepatitis A virus was selected for this test case as the US has recently been experiencing outbreaks of HAV strain 1B.
- Metagenomic/Metatranscriptomic sequencing of emerging pathogens from human specimens. Metagenomic or metatranscriptomic sequencing permits the sequencing and characterization of taxa for which no reference genomes are available. We will apply and optimize metagenomic and metatranscriptomic protocols previously honed on our analysis of microbiome profiling protocols from human-derived samples. For this purpose, we will use human serum, stool, and buccal samples, and in parallel apply the technologies to wastewater samples.
Outcomes:
Overarching outcomes will be the development of robust, high-quality, scalable, and deployment-ready wet-lab methods that can be adopted for PH use. These methods will:
- Improve efficiency, reduce cost, and demonstrate the utility of targeted amplicon sequencing using the PVASP for characterizing HAV in clinical samples.
- Demonstrate the utility of MGS/MTS to identify and obtain draft genomic sequences of HAV in samples from symptomatic individuals.
- Once optimized on HAV, the PVASP and metagenomic/metatranscriptomic technology developed will be applied to other viral pathogens, including but not limited to respiratory and gastrointestinal viruses.
The longer-run plan is to reduce barriers to method implementation within PHLs by providing a demonstration of the rapid development, evaluation, optimization, and validation of standardized methods.
Background: The recent and rapid expansion of NGS within PH has led to a lack of standardization and the need to create robust protocols for the generation of comparable data. Most PHLs employ Illumina short-read sequencing, a method with >99% accuracy that can identify short sequence changes such as SNPs but is limited in identifying large rearrangements and resolving extrachromosomal genetic elements, like plasmids. Long-read sequencing (LRS) technologies such as PacBio and Oxford Nanopore Technologies (ONT) can address these challenges by both improving the assembly of highly repetitive sequences and identifying genome macro-rearrangements, important for tracking the spread of AMR genes. In contrast to Illumina, laboratories have been slow to adopt LRS platforms given the lower accuracy and lack of quality parameters and standard operating procedures (SOPs). The VA PGCoE plans to use our current expertise to perform a multisite assessment of LRS wet lab approaches with the intent to refine and standardize methods for increased adoption in PHLs for PH genomics.
Purpose: To assess the influence of LRS wet lab parameters on the variability of genomic read data across the PGCoE. We will evaluate the ability to close genomes, resolve extrachromosomal structures, and accurately detect AMR genes. This information will identify and resolve inconsistencies that limit the widespread use of LRS platforms in healthcare and PH outbreak investigations.
Outcomes:
- Improve the efficiency in the characterization of HAI/AMR outbreaks by providing evidence-based recommendations for the application of diverse sequencing platforms.
- Establish optimal wet lab approaches to bacterial sequencing to close genomes, recover mobile elements and accurately identify AMR genes.
- Develop standards for optimized wet lab preparations across PHLs.
- Facilitate increased adoption of new sequencing approaches and platforms including the development of quality standards and proficiency testing (PT) materials.
Bioinformatics Technologies
Bioinformatics is the science of analyzing complex biological data, such as genetic sequences. In Genomic Epidemiology, the genetic sequence (DNA, RNA) of a pathogen can be analyzed and compared to sequences from other pathogens to help in identification, paths of transmission, mutation rates, and likely disease outcomes. Click on the links below to learn about bioinformatics techniques in use and development in the VA PGCoE:
Background: Significant effort has gone into developing tools and pipelines for genomic monitoring of the SARS-CoV-2 (SC2) pandemic. We outline strategies to adapt some of these methodologies to develop tools for surveillance of Hepatitis A virus (HAV) in Year 01 and Adenovirus (AV) in Year 02. We adapt the protocols used to classify SC2 to classify variants of HAV and AV. Second, we adapt protocols we currently use to analyze metagenomic sequencing (MGS) and metatranscriptomic sequencing (MTS) data to characterize viral variants. The latter technology has the power to identify any pathogen and to sequence unknown pathogens. Finally, we explore informatics strategies to get genomes of less abundant organisms with informatics approaches and MGS or MTS data.
Purpose: To identify and develop the bioinformatic capabilities to analyze the output generated from targeted genomic sequencing and genomic epidemiological tracking of known pathogens, beginning with HAV. Within this overarching goal, the best practices will be disseminated across the PGCoE infrastructure and tested, and their value will be demonstrated.
Outcomes: The overarching outcomes will be the development of robust software tools to support the application of MTS and MGS to analysis/typing of viral genomes.
- Software paralleling that is in use for the SC2 will be democratized for application to other pathogens, beginning with HAV.
- Pipelines for analysis of MGS and MTS data, to process sequence read data from the formats coming off the in-lab sequencers or sequencing centers through quality filters to reference-based alignment and variant calling, will be established based on currently available off-the-shelf and in-house strategies.
- Sample identifications will be linkable to patient or collection metadata, or anonymized approximations, as appropriate authentication levels permit.
- Pipelines will be disseminated to the Division of Consolidated Laboratory Services (DCLS)/UVA for evaluation and harmonization.
Background: Pathogen whole genome sequencing (WGS) is being increasingly used by clinical and PHLs for pathogen identification, AMR determination, and disease outbreak investigations. Genomic surveillance of an emerging pathogen allows us to build evolutionary phylogenies that can estimate pathogen emergence, characterize its geographic spread, identify instances of adaptation, and track emerging variants, as demonstrated by the viral WGS of SC2 during the current COVID-19 pandemic. While whole genome level inspection provides better resolution into pathogen genomics, compared to traditional PFGE / gene-based multi-locus sequence typing, several challenges remain in its wider implementation -- in particular the standardization and validation of semi-automated pipelines, sequence data quality control, and bioinformatics analysis, computing infrastructure as well as result interpretation.
Purpose: We will demonstrate the ability to quickly determine the genome of an isolated bacterial pathogen on a timeframe relevant to initiating genomic surveillance during an unfolding outbreak. A genetically close reference, preferably at the earliest stages of the outbreak, is important for identifying true phylogenetically informative mutations. This will be then compared to other approaches such as core genome and k-mer approaches to understand differences in resolution. We will finally compare the computational demands of SNV-based whole genome phylogeny approach to further inform when and how it should be employed.
Outcomes:
- Develop (semi-)automated whole-genome SNV-based bioinformatics analysis pipeline(s) for common nosocomial pathogens.
- Validate the SNV-based phylogeny pipeline for nosocomial species and compare to other approaches for false relationships as well as documenting advantages over other comparative approaches.
- Make the comparative genomics analyses underlying the justification of the selection of the final reference available within the PGCoE web portal. This will include phylogenetic trees to show the span of isolates occurring within the outbreak.
Data Standardization, Linkage, Analytics, Visualization
Data is central to epidemiological analysis and response. The VA PGCoE is developing tools and supporting computational infrastructure to enable researchers and responders to analyze and compare, and summarize their data for use in regional and national public health planning and response. Click on the plus sign (+) to expand the list of items below for more information:
Background: Virginia Public Health (PH) entities have identified gaps in lab and epidemiological data integration while responding to the SARS-CoV-2 (SC2) global pandemic. Efforts to address some of these challenges resulted in the establishment of VAS3, a partnership between Virginia academic research centers and the state’s public health laboratory, DCLS. As part of this network, VAS3 partners developed a framework for reporting sequencing data in formats that were HL7-compliant to ease data integration into the Virginia Electronic Disease Surveillance Systems (VEDSS). VEDSS uses the National Electronic Disease Surveillance System (NEDSS), built and maintained by the CDC, to process, analyze, and securely share data on reportable conditions. To increase the portability of sequencing data between VAS3 partners, Amazon Web Services (AWS) Simple Storage Service (S3) was procured, and APHL’s AIMS+ team is currently facilitating the onboarding of partners to transmit sequencing data securely to S3. The VAS3 initiative led to an increase in SC2 sequencing volume (reported to national databases such as NCBI and GISAID) and overall data generation (SC2 lineage incorporation into VEDSS). However, established systems such as VEDSS and LIMS still lack the functionality to meaningfully integrate NGS with epidemiological data. The VA PGCoE is poised to capitalize on existing collaborative infrastructure and groundwork by VAS3 in the form of MOUs, and data use and reciprocal sharing agreements (DURSAs) to expand sequencing beyond SC2 (as laid out in Response Base Section I and III). Currently, there is not a well-provisioned system in PH that allows for easily accessible, secure management and analysis of all genomic, epidemiologic, clinical, biological, environmental, geospatial, behavioral, social, and other types of data together. However, VDH is currently in the process of migrating data management to an enterprise data and analytics portal (EDAP), a Google Cloud Platform (GCP)-based entity. Partners will soon be able to easily access epi data with IRB-approved data-sharing agreements under the Commonwealth’s data trust. Under this new model of data access, a platform could reasonably be piloted to securely accept metadata from the GCP entity and link it to sequencing data for use in downstream visualization, modeling, and analysis.
Here we define an approach for leveraging computational modeling infrastructure to increase detail, connections, and PH inferences through genomics/metagenomics. We plan to bring multi-modeling approaches to the integration of methods and tasks in the project: arriving at standards, best practices, and prototypes for the lab, bioinformatic analysis, biosensing strategies, and epidemiological inferences with models capturing performance and uncertainty at different levels to better facilitate method development and achieve a vertically integrated picture of information and uncertainty given by genomic epidemiology. Data sharing, standardization, and a pervasive computing framework are an integral part of modern surveillance systems. Detailed data models can integrate epi, genomic, and evolutionary data, building a picture of the spatio-temporal-demographic dynamics of pathogens. Unfortunately, current genomic surveillance efforts are fragmented and lack standardization, and pipelines to the extent they exist are not connected to epidemiological modeling and PH decision-making. An integrated data and computing architectural framework is needed to address this important and vexing problem. Building such a system would need to overcome the following challenges: a) tiered access to confidential and sensitive data; b) harmonizing and linking legacy surveillance pipelines; c) developing analytical pipelines that can automate the process that begins with data collection and ends with conceptually organized data structures; and d) integrating genomic data with individual-level clinical, demographic, and behavioral data. The system should scale gracefully and support decision-making by creating rich services; in other words, it should lead to enhanced human productivity. The system should be reliable, interoperable, scalable, and extensible.
Purpose: It is important to build and maintain a platform capable of securely housing NGS data, linking NGS data to relevant epi data, and performing standardized bioinformatics, epidemiological, and biosurveillance analysis to create reports, figures, models, and data analytics for faster, timelier, and more insightful data usage in public health contexts. This platform would interact with existing data repositories to enable seamless data integration between lab and epi sources in GY1-GY2 (figure below) and would be expanded in conjunction with other PGCoEs in GY2-GY5.
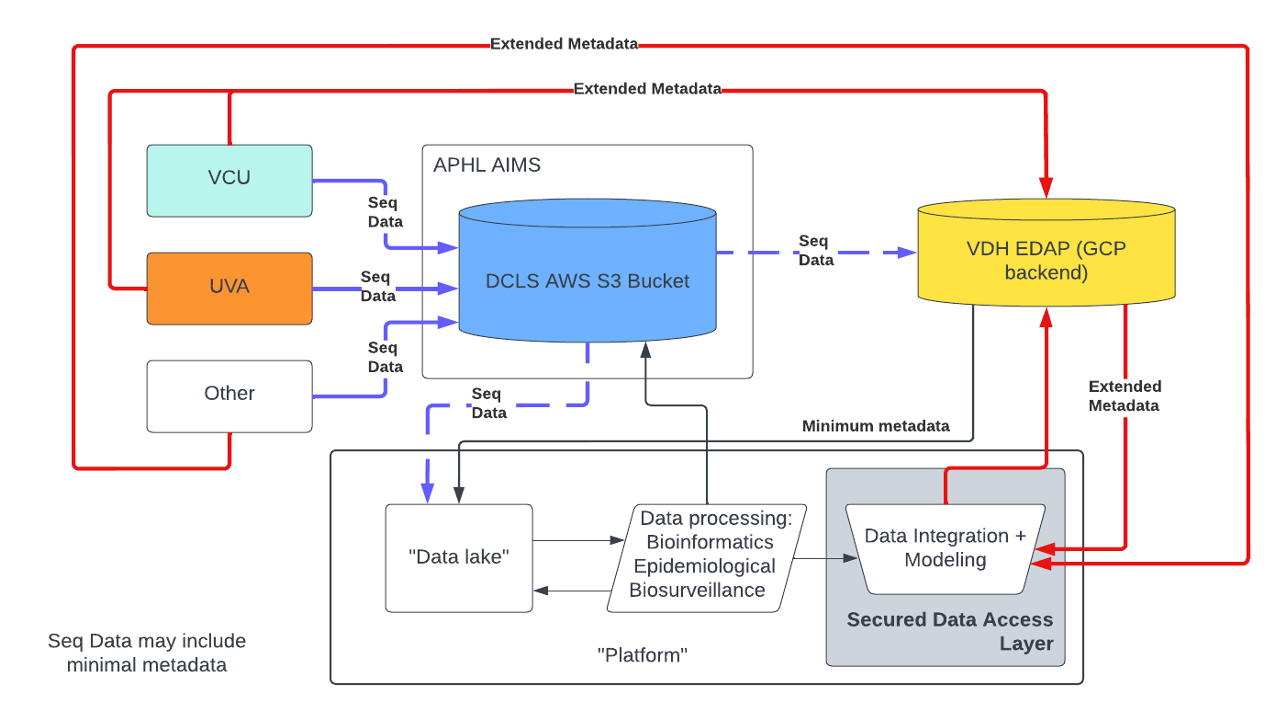
This integrated system would provide analysis for minimizing disease impact, informing interventions, and promoting human health and safety. To enhance and discover analytical methods that describe the relationship between contagion genetics and societal stimuli, data models will be built in an open-world refinement process and used to identify potential public health actions. Such a system requires scalable sensing and computing, genomics, epidemiological models, and bioinformatic signals analysis. We will extend our current capabilities in a refinement process that uses strategic sequence collection as well as existing samples to detect and deconstruct forces that govern the fitness of circulating pathogens.
Outcomes: We expect to achieve the following tasks as part of the Center’s creation:
- Detailed requirements and an in-depth understanding of varied computing and data infrastructures that are currently in use at PHLs regionally and beyond in collaboration with other PGCoEs, designed to identify common components, constraints, and data sharing and governance protocols.
- A novel approach to organizing varied data using the concept of “digital twins” that can make access to data in a tiered manner easy to manage. It will also provide a mechanism to link diverse social, environmental, epidemiological, and pathogen data sets.
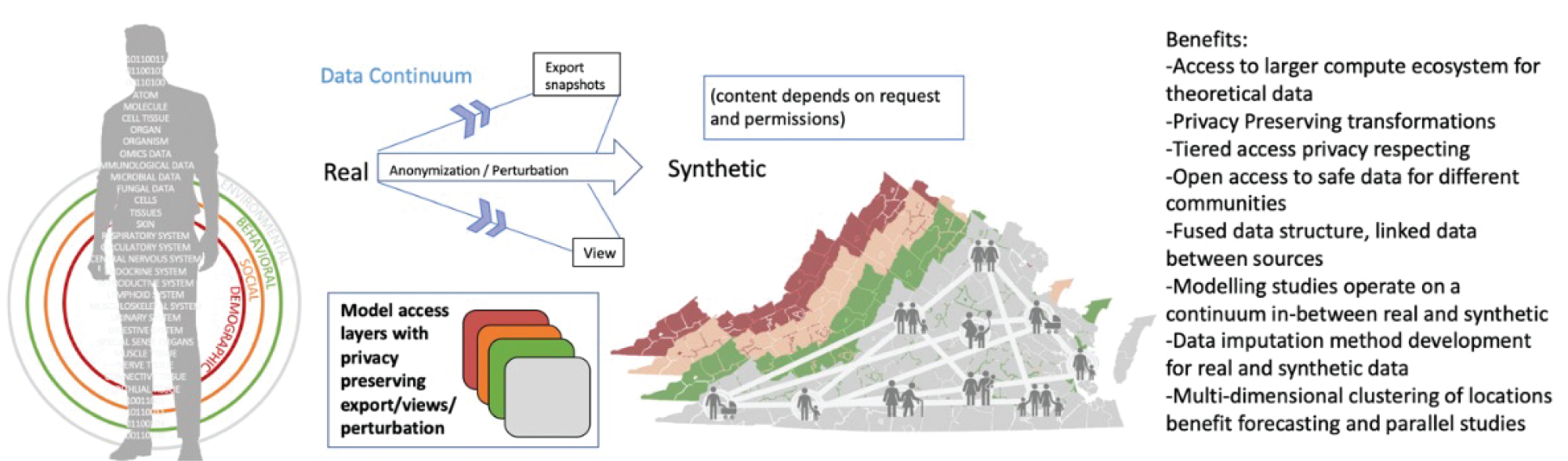
Synthetic model of individuals (digital twin of an individual) and the VA population can be either real associated information or statistically similar information that preserves elements of privacy depending on access privileges or the requested view. One can choose to expose data to the end user at the appropriate level of accuracy and fidelity while still achieving computational parity between the two. - An abductive loop-based approach for smart surveillance that uses high-performance computing (HPC)-based epidemiological modeling combined with techniques in machine learning to guide when, where, and how much to sample to detect weak genomic signals amidst a noisy background.
- A scalable cyberinfrastructure, SCIGS, for seamlessly managing data as mentioned above, workflows for analytics, and web apps.
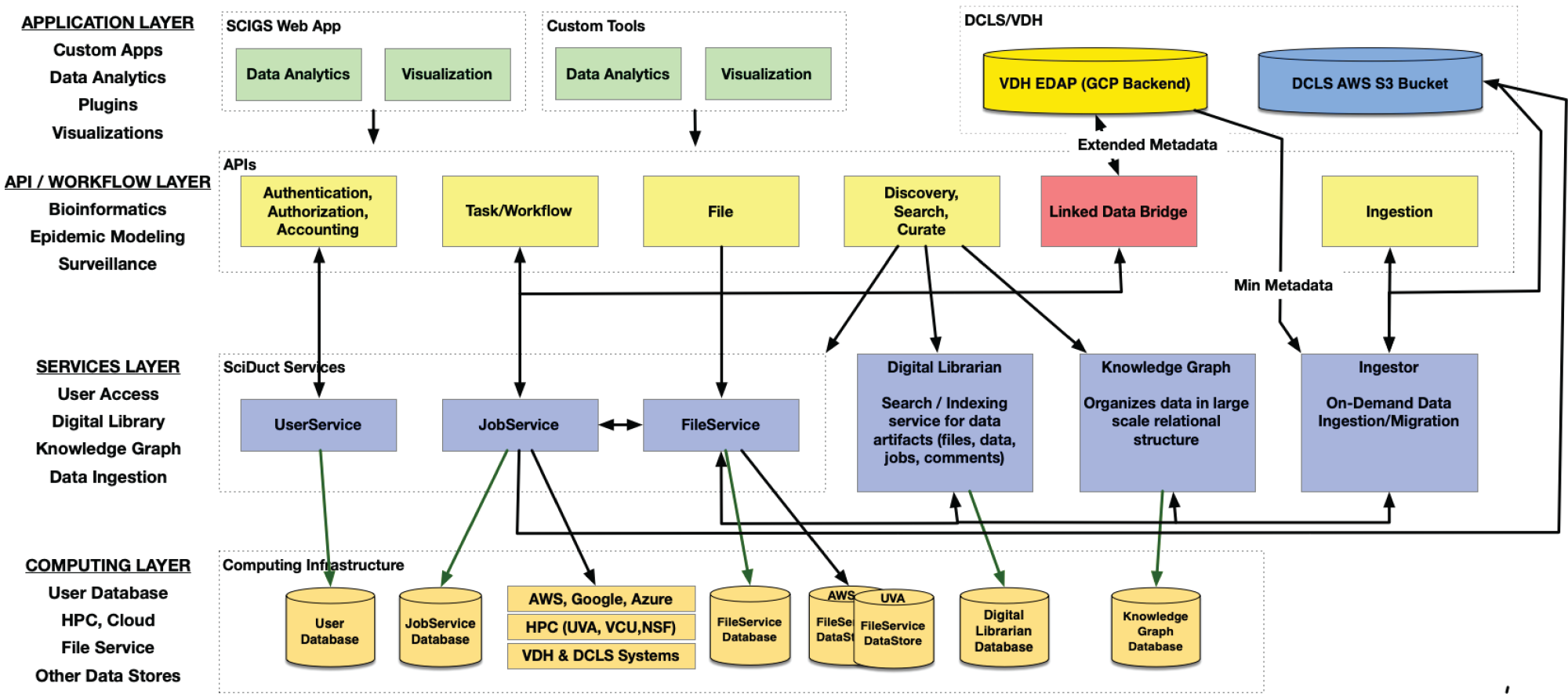
Conceptual layered and privacy-sensitive tiered-access SCIGS system architecture. - Integrated NGS data from all organism and environmental sampling with metadata from health department contexts and investigations. Metadata will include linking identifiers that will carry forward hierarchical relationships among sequences, e.g., multiple samples from the same individual, and multiple sequences from the same sample.
- Established user access protocols to appropriate metadata for report generation and analysis based on security access level.
- Standardized common workflows, pipelines, and data analysis features.
- Use of best practices regarding containerization and workflow management, and tools/software from existing platforms for consolidated analytics.
- Models for better prediction of public health measures utilizing real-world data.
- A platform to easily access and analyze pertinent public health data for making decisions, conducting investigations, and reporting outcomes/trends.
Genomic Surveillance
Genomic Surveillance refers to the monitoring of presence and distribution of pathogen genome sequences collected from clinical patients, public repositories, and passive collection efforts such as wastewater sampling. Epidemiologists use this information to estimate prevalence, spread, and disease risk in populations in near real-time, facilitating impactful response and interventions. Click on the plus sign (+) to expand the list of items below to learn about the advanced methods in practice and development in the VA PGCoE:
Overview: In conjunction with the Field Studies (see sections below on Developing Model for Plasmid Transmission based on Real-World Field Application, Development of Models for Strategic Genomic Surveillance Deployment, and Landscape Analysis for Genomic Surveillance), at roughly six months after gap analysis, method establishment, and assessment of available metadata and diversity of locations available for wastewater (WW) collection, we will proceed with rolling out the project to 24 sites based on metadata resolution, consistency in WW collection as well as ability to capture community diversity. Twenty-four sites will be needed to evaluate the number of unique factors that could influence antimicrobial resistance (AMR). Untreated primary influent will be collected for analysis across all sites. We will compare species-level diversity across sites with alpha and beta diversity. We will also assess both the density of Enterobacterales as well as Candida species. After normalizing, we will compare relative abundances of carbapenemase genes in the context of Enterobacterales density as well as C. auris in the context of Candida density. We will then compare the metagenomic differences to CRE and C. auris culture results and ddPCR for validation. Normalized CRE and C. auris in the context of healthcare factors including National Healthcare Safety Network (NHSN) antimicrobial usage data for hospitals in the sewershed will be evaluated along with other factors such as urban density and residential factors for variation across communities. We will assess the stability of the alpha and beta diversity over time overlaid with factors such as average monthly temperature. Ultimately having this type of surveillance data could be overlaid with clinical AMR data for further validation and could be used to inform the type of antimicrobials that may be empirically prescribed as all the novel agents have variable activity against differing carbapenemases. Developing linked AMR surveillance is critical to developing interventions and WW surveillance offers a community surveillance approach.
Background: During the SARS-CoV-2 (SC2) pandemic the interplay between viral mutations, human behavior, vaccines, and public policies has been unprecedented. An integral element of managing such a pandemic is biosurveillance: collecting samples and sequences related to viruses across space and time, and combining this information to assess the distribution and impact of the viral strains. This is challenging as it depends on the mutation rate, selection pressures, and spatiotemporal data collection. Models for disease surveillance must consider the mode of transmission, budget constraints, resource constraints, disease dynamics, sensor type, and performance profile to optimize detection time, resolution, uncertainty, and cost.
Purpose: Development of models for strategic genomic surveillance deployment.
Outcomes:
- Develop methods to detect the emergence of concerning mutations/strains in SARS-CoV-2.
- Develop computational methods that consider genomics, human behavior, and immune status for detecting or quantifying disease burden in a population.
Field Applications
Field applications are methodologies, procedures, and technologies that are sufficiently developed and standardized to be used day-to-day practice by researchers and public health workers. These technologies yeild proven results that can be used directly for public health planning and response. Click on the plus sign (+) to expand the list of items below for more information on field applications in development in the VA PGCoE:
Background: Some of the most consequential antimicrobial-resistant (AMR) genes are primarily spread between bacterial species by horizontal gene transfer (HGT) of mobile genetic elements (MGE), often carried on self-replicating plasmids. Despite their role in resistance emergence and spread, plasmid behavior remains under-researched, as most efforts focus on the core genomics of the bacterial strain. Sharing of resistant plasmids between bacterial strains is poorly understood, as most genomic sequencing efforts have relied on short-read sequencing technologies, making the assembly and recovery of plasmid-associated AMR genes and their emerging variants challenging. Our long-standing research efforts, since 2007, have involved genomic characterization to understand the transmission of a plasmid-mediated outbreak of multiple Klebsiella pneumonia carbapenemase (blaKPC) producing organisms within hundreds of patients and the hospital environment, using a combination of short-read and long-read sequencing technologies. This ongoing field application presents a tremendous opportunity to understand the evolution of resistant plasmids and develop plasmid-focused genomic tools and applications to understand the basis of dissemination of antimicrobial resistance.
Purpose: To develop plasmid-focused genomic tools to detect multi-species outbreaks that share the same resistant gene for field applications and adoption by the wider Public Health Laboratories (PHLs).
Outcomes:
- Complete long-read and short-read sequencing of both patient and environmental isolates that share plasmids across multiple bacterial species and strains.
- Bioinformatic characterization of plasmid SNVs and indels to estimate factors affecting rates of genomic changes.
- Begin to build mathematical models of plasmid transmission within the hospital from the last 15 years.
Background: Wastewater (WW) surveillance has significantly expanded during the pandemic, and the Division of Consolidated Laboratory Services (DCLS), in coordination with the Virginia Department of Health (VDH), is testing 25 sites weekly for SARS-CoV-2 (SC2) surveillance. DCLS is also piloting WW sequencing for estimation of variant proportions in communities. UVA has a long-standing record of accomplishment in AMR detection in WW including development and validation of genomic techniques. This experience in both laboratories can be applied to development of new and advancement of existing WW methods.
Purpose: Develop field investigations to utilize WW to understand contributions to antimicrobial resistance across Virginian communities.
Outcomes:
- Gather metadata on communities and community hospitals that have participated in WW collection for SARS-CoV-2.
- Gather AMR-specific metadata survey for the Virginia WW surveillance network.
- Establish methods for carbapenem-resistant Enterobacteriaceae (CRE) and C. auris surveillance in WW at a single site and compare results from metagenomics, quantitative digital-PCR, and culture-based analysis.
- Utilize developed methods and sampling network (for WW processing at 25 sites around the state with partners pre-engaged in SC2 sampling) for AMR pathogens: CRE and C. auris and align metadata across sites to compare/refine monitoring and data analysis needs.
Education
Genomic Epidemiology is rapidly evolving, as the capacity and technologies have grown rapidly to collect, share, and analyze genomic sequence data associated with outbreak events. To keep pace with these capabilties, the VA PGCoE is engaged in ongoing education opportunities geared towards researchers and public health workers. Click on the plus sign (+) to expand the list of items below to learn more:
Advanced Training Series: UVA BI will create an advanced training series featuring genomic epidemiology analysis resources (tool, database, or workflow) of relevance to public health and researchers in genomic epidemiology. Each topic will include background epidemiological, biological, and genomic principles that are addressed by the resource. It will also include an overview of the resource, pointing out the scope, key features, inputs, outputs, algorithms employed, any limitations on use, and other relevant information. An example use case will drive the demonstration, illustrating how the resource can benefit public health-related analyses and discovery. The format will be a set of short videos made available via the VA PGCoE website and/or other designated locations, such as the NE educational platform.
Workshop (Panel/Speaker) Series: UVA BI will organize a workshop series featuring topics of interest and timely relevance to genomic epidemiology. The intent is to provide a platform for scientists to share and learn about cutting-edge research, and to discuss the challenges and opportunities for future efforts. Workshops will consist of invited speakers and panel discussions.
Data and Tool Cyberinfrastructure: One of the tasks that UVA BI is undertaking as a part of the project is to develop a Cyberinfrastructure to support our network. We will leverage this effort and develop tools to integrate the data sets, lectures, and workshop material within this CI. Specifically, we will develop a website and supporting backend infrastructure with a webpage for each of the data and tool resources featured in the Advanced Training Sessions described above. The page will have information about the resource, links to training information and videos, relevant citations, links to the resource, and/or if appropriate and feasible, an instance of the resource deployed on the website.